Haemophilia
Affecting around 1 in 3500 births worldwide, haemophilia is a disorder cause by a reduction or absence of the clotting factors FVIII (haemophilia A) and FIX (haemophilia B). This reduction or absence means that that the clotting pathway is either impaired or incomplete which leads to inadequate or incomplete clotting in affected persons.
Article by Simon Fletcher
Expand all
Collapse all
Definition
Affecting around 1 in 3500 births worldwide (Stonebacker et al, 2020) haemophilia is a disorder cause by a reduction or absence of the clotting factors FVIII (haemophilia A) and FIX (haemophilia B) (Calaghan and Kaufman 2010, Kitchen et al, 2020). This reduction or absence means that that the clotting pathway (Figure 1) is either impaired or incomplete which leads to inadequate or incomplete clotting in affected persons.
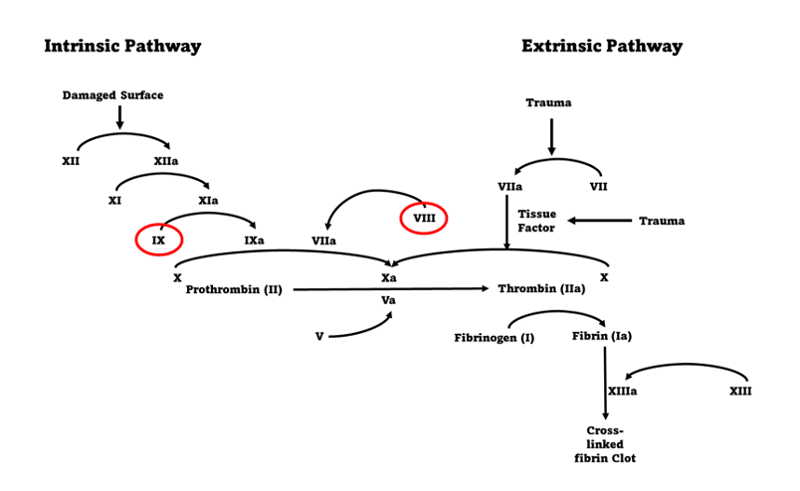
Normal factor levels in children and adults range between 0.5iu/ml and 1.5iu/ml and individuals with a factor level <0.5iu/ml is indicative of haemophilia (Hatton et al, 2018). The disorder is further subdivided into 3 severity types:
- mild (factor levels between 0.05iu/ml and 0.5iu/ml)
- moderate (factor levels between 0.01iu/ml
To view the rest of this content login below; or read sample articles.
Symptoms
Symptoms depend on the severity of the presentation but include, easy bruising, epistaxis, mucosal bleeding, haemarthrosis (particularly in the knee and ankle joints) and muscle bleeds (Proven et al 2009; Khair et al 2014; Hanley et al, 2017).
If untreated, people with severe haemophilia have frequent, unprovoked joint and muscle bleeds, some of which can be life threatening (Lee, 2010).
Men with mild and moderate haemophilia, as well as carrier women with low factor levels, tend not to suffer spontaneous bleeds but will have prolonged bleeding if injured or require surgery.
To view the rest of this content login below; or read sample articles.
Complications
One third of people with haemophilia can develop auto antibodies to the factor products they take to prevent and or treat bleeds, negating its effectiveness. People with haemophilia and inhibitors are therefore unable to use factor products to treat any bleeds they may have and have to use bypassing agents (FVII or activated Prothrombin Complex Concentrate (APCC)) (Santagostino et al, 2009; Oldenburg et al 2017; Ragni et al, 2020).
To view the rest of this content login below; or read sample articles.
Aetiology
Haemophilia is an X linked recessive disorder of the gene that codes for the production of either FVIII or FIX. In approximately two-thirds of presentations the gene is passed from carrier women to their sons. However, one-third of presentations are caused by mutations which occur de novo, and there is no prior family history of haemophilia.
The mutation can also be passed from affected men to their daughters who will be obligate carriers of haemophilia and can in turn pass the condition on to their children (Calaghan and Kaufman 2010). Boys who inherit the mutation will always be affected but girls will (except for in some very rare cases through skewed lyonisation or inheritance of a genetic mutation for severe haemophilia on both X chromosomes) be carriers (Shoukat et al, 2020; Miller and Bean, 2021; Foji et al, 2021). This does not mean that women do not suffer with many
To view the rest of this content login below; or read sample articles.
Diagnosis
In families with a known history of haemophilia, diagnosis is normally made during pregnancy though confirmatory testing can be undertaken at birth if umbilical cord blood is taken and tested.
If there is no family history of haemophilia, then diagnosis is normally made in infancy or early childhood, depending on the severity of the condition. In this case a clotting screen can be performed, which would show a prolonged activated partial thromboplastin time (APTT). Subsequent testing for FVIII and FIX will show which of the two factors is deficient and to what extent (Hatton et al, 2018; Kitchen et al, 2020).
Carrier girls are not normally tested until school age, when their factor levels can be checked. Genetic testing would not normally be done until they are old enough to consent for the process themselves (Dunn et al, 2008). They will then be followed up by their local haemophilia care
To view the rest of this content login below; or read sample articles.
Treatment
For many years there was no treatment available for haemophilia other the transfusion of whole blood and or cryoprecipitate (Lee, 2018). As the individual clotting factors were identified and fractionated, they were developed initially as a treatment for bleeding episode but eventually began to be used prophylactically to prevent bleeds form occurring. There are now a growing number of treatments available for people with haemophilia.
Factor replacement
Pooled Plasma Factor Products
Pooled plasma products are available and are now virally inactivated significantly lessoning the risk of transmission of viral pathogens (Pipe et al, 2020). They are not widely used in the UK (UKHCDO, 2021).
Recombinant Factor Products
These are synthetic factor products and as such have no known risk of viral contamination.
Both pooled plasma and recombinant factors are given intravenously. Both can be given regularly to prevent bleeds from occurring and/or intermittently to treat bleeds that do occur
To view the rest of this content login below; or read sample articles.
Resources
References
Baty P, Lillicrap D. Advances and Challenges for hemophilia gene therapy. Human Molecular Genetics. 2019;28:R95-R101 https://doi.og/10.1093/hmg/ddx1547
Calaghan MU, and Kaufman RJ. Cellular processing of factor VIII and factor IX. In: Lee C, Berntorp E, and Hoots K (eds). Textbook of Haemophilia. (2nd edn). Oxford, UK: Wiley-Blackwell: 2010. p 13-23
Carcao, M, van den Berg HM, Gouider E et al. Prophylaxis in Hemophilia. Haemophilia. 2020;26(S6):72-84.
Chowdary, P. Anti-tissue factor pathway inhibitor (TFPI) therapy: a novel approach to the treatment of haemophilia. Int J Hematol. 2020;111:42–50. https://doi.org10.1007/s12185-018-2548-6
Collins PW, Liesner R, Makris M et al. Treatment of bleeding episodes in haemophilia A complicated by a factor VIII inhibitor in patients receiving Emicizumab. Interim guidance from UKHCDO Inhibitor Working Party and Executive Committee. Haemophilia. 2018;24:344–347. https://doi.org/10.1111/hae.13495
Dunn NF, Miller R, Griffioen A, Lee CA. Carrier testing in haemophilia A and B: adult carriers’ and their partners’ experiences and their views on the
To view the rest of this content login below; or read sample articles.